Introduction
The grass family (Poaceae) can be largely divided into two clades which are the BEP (named based on the first letters of subfamilies Bambusoideae, Ehrhatoideae, and Pooideae) and the PACCAD (the first letters of subfamilies Panicoideae, Aristidoideae, Chloridoideae, Centothecoideae, Arundinoideae, and Danthonioideae) clades. The BEP clade represents C3 photosynthetic system grasses whereas the PACCAD clade represents C4 photosynthetic on es. C4 photosynthetic ones. The BEP clade including rice and small grains are major food crops for humans while the PACCAD clade represented by maize, sorghum, and millets are used for both forage and food crops. The physiological differences between these two clades result in efficiency differences between the groups’ light, water, and nitrogen use, as well as in their plants’ biomass digestibility and decomposability.
The advantages of the C4 photosynthetic system have frequently been reported. The elevated level of carbon dioxide (CO2) concentrations in the atmosphere has become a major issue since the 20th century. High CO2 levels have caused climate changes that may have a dramatic impact on agricultural conditions. In fact, the elevated concentrations of atmospheric CO2 do not reflect the low CO2 environment under which C4 photosynthesis has evolved. Nonetheless, photosynthetic nitrogen (PNUE) and water (PWUE) use efficiencies were highly maintained in C4 species under high CO2 conditions (Pinto et al., 2014), indicating that the C4 photosynthetic system is superior to the C3 system even under the current global warming conditions. A key feature of C4 photosynthetic systems resides in the condensation of atmospheric CO2 as a form of 4-carbon organic acids (malate). The CO2 released from the malate is fixed by Rubisco for further reaction through the Calvin cycle. The 3-carbon organic acid (phosphoenolpyruvate) that released CO2 accepts new CO2 through light-dependent reactions to form malate. This condensing process increases water use efficiency up to 40% compared to the C3 photosynthetic system (Taylor et al., 2014) owing to higher photosynthetic rates per unit leaf area and a lower stomatal conductance. Therefore, C4 systems are adapted to high light intensities, high temperatures, and dryness. In the same aspect, C4 grasses are usually predominant in the tropical and subtropical regions compared to C3 grasses (Edwards et al., 2004).
The grass subfamily Panicoideae includes over 3,500 species in 206 genera (Grass Phylogeny Working Group, 2001). This subfamily is of particular importance in that it includes a plethora of economically important C4 crops such as maize (Zea mays), sorghum (Sorghum bicolor), sugarcane (Saccharum officinarum), common millet (Panicum miliaceum), pearl millet (Pennisetum glaucum), foxtail millet (Setaria italica), Miscanthus spp., and switchgrass (Panicum virgatum) . Although this subfamily consists of 13 tribes, two tribes, Paniceae and Androporoneae, are the most noticeable in terms of the number of species included (Simon, 2007). In particular, some major cash crops such as maize, sorghum, Miscanthus, and sugarcane belong to the tribe Andropogoneae, which is further divided into several major subtribes such as Tripsacinae (maize), Sorghinae (sorghum), and Saccharinae (Miscanthus and sugarcane). Grasses included in the Genera Saccharum (sugarcane), Miscanthus, Erianthus, Narenga, and Sclerostachya form an interspecific breeding group called the Saccharum complex, within the Andropogoneae tribe of grasses (Daniels et al., 1975). In the aspect of plant genomics research, the Saccharinae group of grasses with its several recent genome duplications may provide a fascinating system to offer insights into the many diverse consequences of this widespread evolutionary process (Kim et al., 2014b).
Maize is used for food and forage, and as feedstock for 1st generation bioethanol production. It leads US crops with a farm-gate value of $15-20 billion per year. S. bicolor, native to Africa with many cultivated forms nowadays, is an important crop worldwide. It is used for the production of food, forage, alcoholic beverages (mostly in China), and biofuels (a substitute for maize). Sorghum may be the most heat- and drought-tolerant crop in the grass family, reflecting its importance in arid regions. Sorghum varieties form important components of pastures in many tropical regions. S. bicolor is also an important food crop in Africa and South Asia, which is the "fifth-most important cereal crop grown in the world" after maize, wheat, rice, and barley (http://www.fao.org/). Sugarcane is an important crop worldwide, producing about 80% of the world’s raw sugar and is getting more attention as feedstock for the production of 1st generation bioethanol (de Setta et al., 2014). The genus Miscanthus has also been considered attractive as feedstock for lignocellulosic biofuel (2nd generation bioethanol) production because it produces high yields of biomass but with low-nutrient requirements, adapts well to marginal land with resistance to abiotic stress, and does not compete with use for food.
In this article, genomics research using reference genomes in the tribe Andropogoneae is intensively reviewed, emphasizing the Saccharinae complex. Besides, the use of genomics tools for various breeding procedures is briefly covered. Despite the economic importance of the Saccharinae complex, genomics studies have lagged far behind mostly due to their large genomes and high ploidy numbers. However, close reference genomes such as maize and sorghum have accelerated the unraveling of those complex genomes with a variety of comparative genomics approaches. Advanced genomics technologies will make it possible to manipulate and control important traits in the future.
Reference genomes of Andropogoneae
We cannot overemphasize the importance of maize and sorghum as cash crops. However, there are also many reasons and advantages to using maize and sorghum as model species for genomics study. First, they have relatively simple genomes despite the high content of repeated sequences in maize. They are diploids and display disomic inheritance patterns with self-pollinated reproductive systems. Second, their closeness to some complex genomes might provide another opportunity to investigate sugarcane and Miscanthus. Last, the rich history of their genetics made things much easier for genomics approaches for these species. Therefore, their genome projects started long before the next-generation sequencing (NGS) technologies became popular. Since Arabidopsis genome was first sequenced in the Viridiplantae (Arabidopsis Genome Initiative, 2000), rice (Oryza sativa L. ssp. Indica and Japonica) was the first grass fully sequenced (Goff et al., 2002; Yu et al., 2002). However, rice is a C3 grass which diverged from C4 grasses approximately 50 million years ago (MYA) (Kellogg, 2000). Consequently, it is not reliable to use the rice genome as a reference for maize or sorghum. The completed genomes of maize and sorghum have been intensively used as reference genomes in the tribe Andropogoneae as well as other C4 grasses (Paterson et al., 2009; Schnable et al., 2009). These reference genomes provided the basis for understanding the C4 photosynthetic system by profiling C4-specific genes as well as their evolutionary history like the domestication process.
Comparative genomics approaches for larger-genome species
Since the Saccharum complex is characterized by its complex and high ploidy genomes, reference genomes are highly useful for genomics research of such complex species. For example, M. × giganteus (2n = 3× = 57), which is specifically of interest due to its ability to accumulate biomass, is thought to result from crosses between M. sacchariflorus (2n = 4× = 76) and M. sinensis (2n = 2× = 38) (Sacks et al., 2013). The genome sizes of M. × giganteus, M. sacchariflorus, and M. sinensis are reported as 6.8 Giga base pairs (Gbp), 4.3 Gbp, and 5.3 Gbp, respectively (Rayburn et al., 2009). The genomic structures of Saccharum spp. are even more complicated. Modern hybrid sugarcane cultivars are interspecific hybrids that typically have 100 to 130 (2n) chromosomes with many chromosomal fragments contributed from S. officinarum (-80% of total) but only -10 to 15% of the genome contributed from Saccharum spontaneum, a wild relative. S. officinarum has been defined by some as having 2n = 80 chromosomes (Daniels and Roach, 1987), consistent with -92% (459) of 497 genotypes classified morphologically as this species for which basal chromosome numbers were x = 10 (Irvine 1999). In contrast, S. spontaneum genotypes vary in chromosome number from 2n = 40 to 128 with a predominant 2n = 64 form, suggesting a basal chromosome number of x = 8 (D'Hont et al., 1998; Ha et al., 1999). The 100 to 130 chromosomes of modern hybrid sugarcane cultivars may originate from interspecific hybridization with restitution of the maternal S. officinarum chromosome complement (Bremer, 1961), together with a high tolerance for aneuploidy (Grivet and Arruda, 2001). Thus, the study of the large genomes of Miscanthus and Saccharum is quite challenging. However, the completed genome of sorghum, which is the closest model species in the same tribe, has provided valuable comparative genomics resources. The sorghum genome has not seen additional genome duplications since the pan-cereal duplication event about 65 MYA (Paterson et al., 2004). However, Saccharinae grasses have formed their current genome structures with multiple duplication events with various crosses.
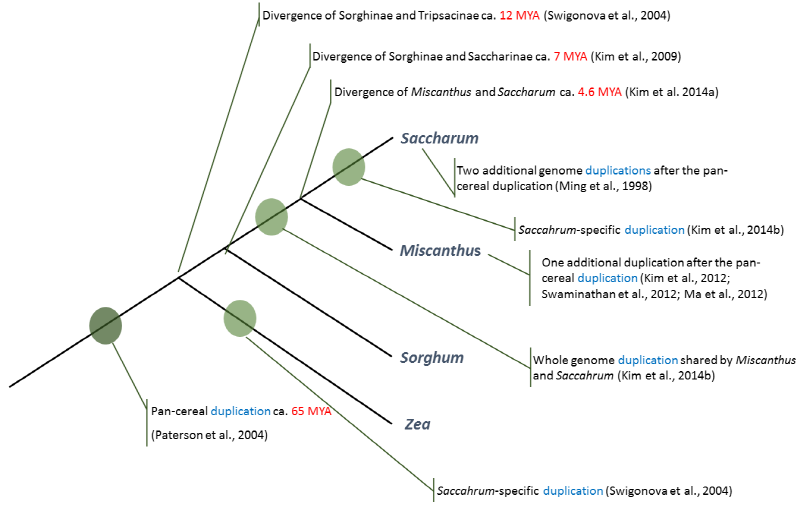
Fig. 1 summarizes the key genomic events to form the current genomic structures of four representative species included in the tribe Andropogoneae. Since the pan-cereal duplication event, the subtribe Tripsaceae (maize) diverged from others about 12 MYA and maize experienced a whole genome allotetraploidization (Swigonova et al., 2004). After that, maize was diploidized to form a current genomic structure, 2n=2x=10. Sorghum, a representative of the subtribe Sorghinae, diverged about 7 MYA from the Saccharinae complex (Kim et al., 2009). As briefly discussed, sorghum has had no additional duplication event and has a relatively compact genome (ca. 0.75 Gbp) compared to other grasses in the same tribe, making this species a good model species to further investigate complex genomes such as Miscanthus and Saccharum. According to previous genetic studies, Ming et al. (1998) reported that Saccharum experienced two additional duplications to form an octoploid genome while Miscanthus had one additional duplication event since the pan-cereal duplication event (Kim et al., 2012; Ma et al., 2012; Swaminathan et al., 2012). Based on that information, paralogous genes in Miscanthus and Saccharum were dated using sorghum genome, indicating that those two species share the whole genome allotetraploidization, which triggered the divergence of the two grasses ca. 4.6 MYA (Kim et al., 2014a) and Saccharum underwent lineage-specific autotetraploidization (Kim et al., 2014b).
|
|
|
Fig. 1. Major genomic changes of four grasses included in the tribe Andropogoneae. Circles indicate the whole genome duplications that occurred during their evolutionary histories. |
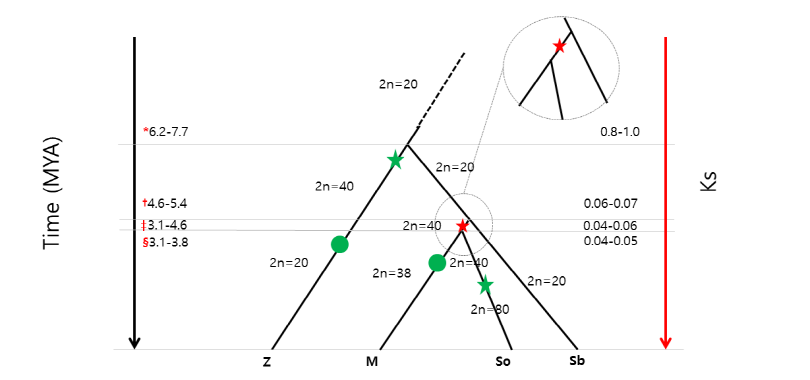
Another interesting point inferred by a series of the comparative approaches is the evolution of basal chromosome numbers of the Andropogoneae. Before the divergence of the BEP and the PACCAD clades, the common ancestor is thought to have 12 chromosomes (x = 12) (Abrouk et al., 2010). The subfamily Panicoideae now has a basal chromosome number of 10 through some genomic rearrangements (Swigonova et al., 2004; Wei et al., 2007). Therefore, sorghum and maize did not have further chromosomal rearrangement since they are consistent with x = 10. On the other hand, Miscanthus has x = 19 whereas Saccharum has a variety of basal chromosome numbers from 8 to 10. Fig. 2 shows the summary of chromosome numbers according to major genomic changes of four grasses in the tribe Andropogoneae. In fact, a high density genetic map using NGS technology inferred the chromosomal fusion in M. sinensis (Swaminathan et al., 2012). They explained the reduction of chromosome number from x = 20 (by the shared allotetraploidization) to x = 19 (the current form) with the insertional dysploid model proposed by Luo et al. (2009) in Aegilops tauschii which is one of the genome progenitors to hexaploid wheat (Triticum aestivum). Therefore, the current genome of M. sinensis experienced another diploidization after the chromosomal fusion shown in Fig. 2.
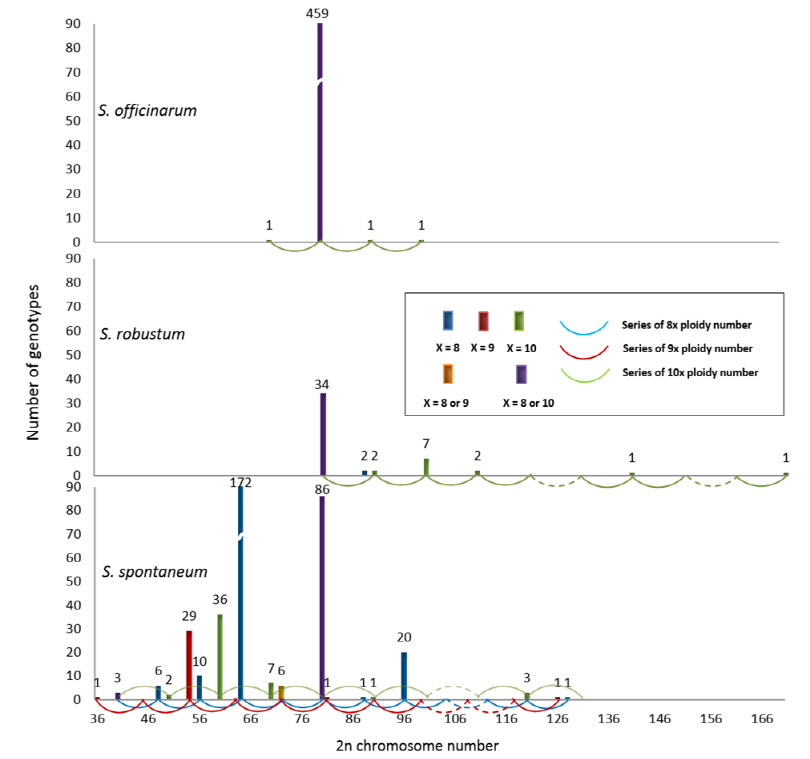
The progression to Saccharum’s basal chromosomal number may be more complicated because of a wide range of variations. Fig. 3 indicates the basal chromosome numbers of various accessions in S. officinarum, S. robustum, and S. spontaneum which are representatives of Saccharum spp. The data was originally collected by Irvine (1999) and reformatted by Kim et al. (2014b). As conventional cultivars with a large amount of sugar production, the genomes of S. officinarum and S. robustum remain quite fixed to x = 10 as shown in Fig. 3. However, a wild species, S. spontaneum, displays many variations in somatic chromosomal numbers. The point is that all the variations can be explained by only two basal chromosomes, x = 8 and x = 10. Although the data indicate the reduction of basal chromosome numbers from x = 10 to x = 8 in this wild species, further investigation is needed to find whether x = 9 is just a transitional form of chromosomal reduction or a natural hybridization between x = 8 and x = 10. Researchers also suggested another hypothesis that further chromosome number reductions have occurred in the Narenga-Sclerostachya clade, another part of the Saccharinae complex that is less represented, with a basal number of 15 (Grivet et al., 2006). In fact, further investigation of genome structural evolution might accelerate the understanding of the phylogenetic relationships among Saccharinae taxa, which have been extremely difficult to deduce and remain uncertain (Kellogg, 2013).
Sugarcane Genome Sequencing Initiative (SUGESI)
Due to the low price of crude oil, bioethanol-related research is less popular than it used to be but sugarcane is still an important crop as a primary source for the sugar industry. As stated, hybrid sugarcane cultivars have very complex genomes which have been a major stumbling block for genetics and genomics studies. However, the Sugarcane Genome Sequencing Initiative (SUGESI) is an international consortium for genome sequencing of this complex species (Souza et al., 2011). Researchers from several different countries (Brazil, US, France, Australia, and India) are extensively involved in the SUGESI and are contributing to the whole genome sequencing of sugarcane using comparative genomics approaches using the reference genome of sorghum. A variety of preliminary studies were conducted to acquire information on the complex genome structure for the whole genome sequencing. For example, a BAC library of hybrid sugarcane cultivar R570 was already constructed (Tomkins et al., 1999). Researchers involved in the consortium started by sequencing BAC-ends to look into the landscape of the hybrid sugarcane (Kim et al., 2013). Fig. 4 shows the comparison between BAC-end sequences (BESs) and the sorghum genome. When low copy BESs were compared to the sorghum genome, they cover about 200 mega base pairs (Mbps), consisting of the entire size of euchromatic regions of sorghum. Those BACs were specifically selected for the initial sequencing of hybrid sugarcane cultivars because of their high possibility of being in the euchromatic regions of hybrid sugarcane cultivars, and these results were published soon after (de Setta et al., 2014). With the advances of NGS technologies, the entire genome has been sequenced with various platforms at this point. Researchers expect that the whole genome draft sequences will be publicly available soon, contributing to the advancement of sugarcane research.
Use of genomics resources for future breeding efforts
Crop breeding is a key element of agricultural research and development. Breeding practices have been carried out since humans started settled agriculture about 10,000 years ago. From conventional selection methods to recent molecular breeding techniques, a variety of efforts have been made to form current crop cultivars and this procedure will continue in the future, especially to overcome several issues such as population increase and climate changes. Conventional breeding methods through artificial crosses and selection largely depend on the subjective experience and visual estimation of crop breeders. However, new methods using molecular markers (marker-assisted selection, MAS) provide an innovative way to reinforce crop breeding pipelines. A molecular marker has many advantages: 1) it is less affected by the environment, 2) theoretically, the number of molecular markers is almost infinite, and 3) it is very objective. Molecular breeding using molecular markers made a remarkable turning point when the complete genomes of Arabidopsis and Oryza were released in the early 2000s. As of 2016, approximately 100 plant genomes including green algae have been sequenced (https://genomevolution.org/coge/) and comparative genomics approaches using these resources allow advances in molecular breeding for ‘orphan crops’ which have scarce breeding resources. For example, the information obtained from model species permits both the rapid identification of candidate genes through bioinformatics analysis and the discovery of molecular markers through comparison of the reference genome with sequence data from closely related species. Particularly, the advent of NGS technologies significantly reduced the cost of genome sequencing, resulting in the frequent use of single nucleotide polymorphisms (SNPs). SNPs have allowed researchers to apply large-scale genetic analyses to various breeding populations and to increase the density of genetic maps, which make it easier to locate quantitative trait loci (QTL) that consist of the most important agronomic traits. Consequently, the development of crop genomics may be meaningful for the increase of agricultural productivity through crop breeding.

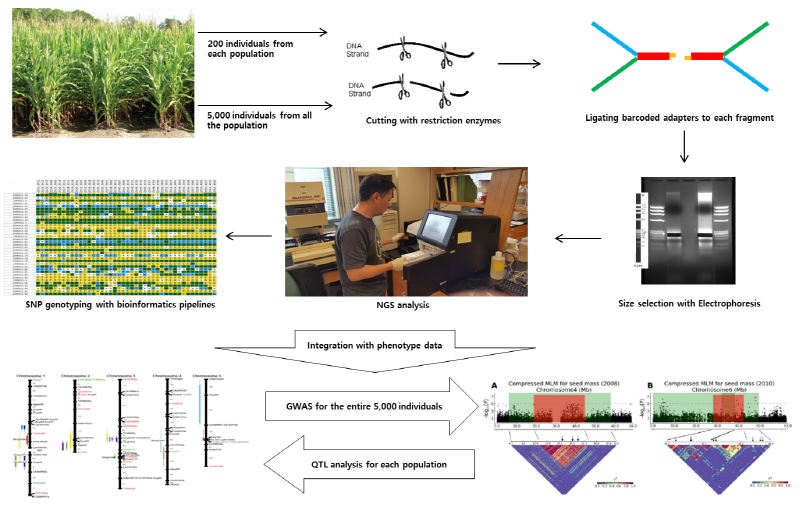
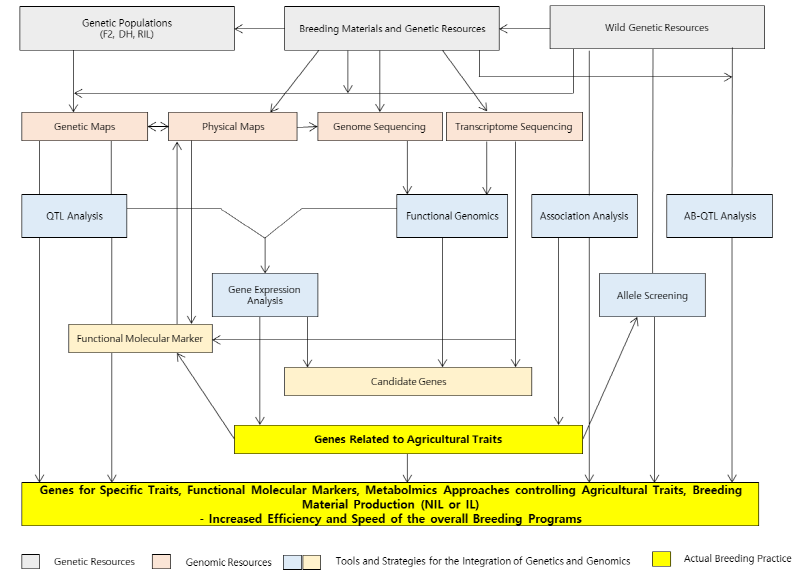
Recently, genomics-assisted breeding (GAB) is getting more attention due to its high efficiency in terms of time, money, and labor. The initiation of a GAB pipeline is not very different from common breeding pipelines. Fig. 5 summarizes the outline of genomics-assisted breeding. The collection of breeding materials such as wild genetic resources and commercial cultivars may be the first step and the establishment of breeding populations will follow. However, to maximize the accuracy and efficiency of selection procedures, genomic information should be collected. For example, high-density genetic maps can be constructed using various crossing populations (F2, DH, and RIL). Whole genome sequencing can be accomplished and the sequencing information can be anchored to those high-density genetic maps in order to locate candidate genes on the physical chromosomes. Besides, transcriptome analysis may provide the expression profiles of important genes. In summary, all the information gathered through different genomics studies can be integrated to produce a synergetic effect with existing molecular data. The advance of crop genomics has also facilitated the studies of wild relatives of important crops due to the reduced cost of mass analyses of genotypes and phenotypes. For example, many genes (especially, ones related to adapting to unfavorable environmental conditions) are lost during crop domestication. However, wild relatives may have more variations and traits than domesticated crops. Thus, various traits from wild species can be introduced to the elite cultivars to increase their adaptabilities to harsh environmental conditions. As stated, NGS technologies have provided opportunities to use SNPs at reasonable costs, making it more cost effective to do research on wild genetic resources with less economic importance. In addition, the advance of bioinformatics tools also permits the processing of big data generated by a variety of genomics research. The representative methods to use wild genetic resources may include association analysis (marker-trait association, MTA) and AB-QTL (advanced-backcross QTL). An MTA analysis correlates phenotypic data to SNPs distributed across the whole genome in order to find candidate SNPs tightly linked to a specific trait. This method enables sophisticated analysis in that it utilizes meiotic recombination information accumulated for hundreds of generations. However, this uses diversity panels which are randomly collected from many wild species or cultivars, causing a number of type I errors (false positives) that result from uncertain subpopulation structures. Using software like STRUCTURE (Pritchard et al., 2000) when examining a subpopulation structure, those errors can be minimized. On the other hand, AB-QTL analysis has been very limited due to complex procedures for its genotyping, but many researchers are revisiting it because of the advances in mass-genotyping technologies. The AB-QTL has evolved to a more complicated method called nested-association mapping (NAM) which is now being adopted by many research groups to discover useful alleles for breeding. Fig. 6 shows an example of mass-genotyping and QTL analysis with NAM populations as breeding materials. This series of analysis integrates some advantages of MTA and AB-QTL procedures because subpopulation structures of starting material can easily be recognized. The integration makes it possible to utilize fine genetic maps as well as association analysis founded based on meiotic recombinations within the NAM populations. The molecular markers tightly linked to target traits may be valuable resources for different breeding purposes. Although sugarcane and Miscanthus have much more complicated genome structures without reference genomes, the same pipeline can be used for breeding those species with the fruitful background information that has been produced in addition to the well-established sorghum genome as a comparative genomic resource.
Conclusion
The subtribe Andropogoneae has attractive features for crop genomics study as well as agricultural use. Their history, rich in major genomic changes, allows researchers to establish academic models for comparative genomics using closely related reference genomes. The genomic resources of Miscanthus and sugarcane obtained from comparative studies may be useful for breeding those complex species. The whole genome draft sequence of sugarcane assembled based on the sorghum genome will provide invaluable information on its supreme capability for sugar production and wide range of chromosomal variations.
NGS technologies have accelerated the genomics research of many plant species. Whole genome sequencing becomes a more common procedure for various goals towing to reduced sequencing costs. Accordingly, researchers have changed their research focuses from major crops to orphan crops as well as to wild species that may have had many useful genes deleted during crop domestication. The evolutionary study of genomic structure has provided the basis for understanding relationships among different species. In addition, high-throughput genotyping using NGS technologies has generated ultra-high density genetic maps which can be utilized to find agricultural traits underlying certain genomic regions. Consequently, in the field of crop breeding, marker-assisted selection, considered innovative about 15 years ago, has evolved into genomics-assisted breeding. Big agro-genomic data being produced owing to these technical advances will be a crucial driving force for crop breeding procedures to be prepared for increased population and climate changes.