
Introduction
포도는 전세계적으로 생산되는 과수 작물 중 하나로, 레스베라트롤 뿐만 아니라 플라보노이드, 폴리페놀계 화합물이 풍부하여 생과, 와인, 건강식품 등 다양한 소비 형태로 소비되고 있다(Reisch et al., 2012). 또한, 국내에서도 2014년도 기준 16,931 ha, 26만톤이 생산되는 주요 과수 작물에 속하며(FAO, 2014), 양질의 포도를 생산하기 위한 많은 유전-육종 연구가 이루어지고 있다. 그러나 포도 역시 일반 과수 작물과 마찬가지로 육종 기간이 길고, 이형접합(heterozygote)의
유전적인 특성을 지니고 있어 육종 연구에 많은 어려움이 따르는 것이 사실이다(Mylesa at al., 2011). 실제로 국내 포도 재배면적의 약 70%를 차지하는 캠벨얼리 품종은 미국 도입 품종으로 전국 각지에서 재배가 가능할 뿐만 아니라 내한성이 강하기 때문에 현재까지도 국내 주요 품종으로 자리하고 있다(FAO, 2014). 최근 ‘흑구슬’이나 ‘흑보석’ 등 국내 육성 신품종의 신규 재배면적이 급속히 늘고 있지만, 우리 고유의 재배종을 비롯해 내재해성을 갖는 새로운 포도 품종 육성이 필요하며, 육종 소재로서의 활용도를 높이기 위한 다양한 유전체 및 분자 표지 마커의 개발이 요구되고 있다.
분자 마커(Molecular marker)는 작물의 유용한 형질을 탐색하거나, 육종 시 개체를 선발하는데 매우 중요한 의미를 가지며(McCouch et al., 2002), 형질 연관 마커 뿐만 아니라 내병성, 내한성 등 다양한 환경 요인에 저항하는 유전자의 선발을 가능하게 한다(Zhang et al., 2014). 최근 NGS (Next Generation Sequencing) 기술이 발달하면서 더욱 쉽고 빠르며 경제성이 있는 마커 개발을 위해 전체 염기서열을 해독하고 결정하는 기술과 고밀도 유전자 지도를 작성하는 기술 및 연구 등의 중요성이 매우 커지고 있는 이유이다(Varshney et al., 2009; Poland and Rife, 2012). 포도는 1995년 최초로 동위 효소와 RFLP (Restriction Fragment Length Polymorphism), RAPD (Randomly Amplified Polymorphic DNA) 표지를 이용하여 병 저항성과 연관된 표지를 개발하기 위한 유전자 지도가 만들어졌고(Lodhi et al., 1995), 그 이후로 AFLP (Amplified Fragment Length Polymorphism), BAC end sequence-기반, EST-기반, SSR (Simple Sequence Repeat), SNP (Single Nucleotide Polymorphism) 등 다양한 유전체 연구가 보고되고 있다(Vezzulli et al., 2008; Hur et al., 2015). 그러나 이러한 NGS 기술을 기반으로 이루어지는 연구는 유전체 전체를 해독함으로써 다양하고 정확한 결과를 얻을 수는 있지만, 대부분 많은 시간과 비용이 발생하며 불필요한 영역을 포함한 방대한 양의 게놈 정보까지 얻게 된다. 결국 이러한 단점이 부각되면서 이를 보완하기 위해 다양한 NGS 기반의 새로운 기술이 도입되고 있다(Poland and Rife, 2012).
옥수수를 소재로 처음 개발된 GBS (Genotyping-By-Sequencing) 기술은 특정 제한효소(ApeKI )를 사용하여 절단된 주변 서열을 중심으로 염기서열을 해독하고 분석하는 기술로서, 기존의 NGS와 달리 분석 비용이 저렴하다. 또한, 바코딩 시스템을 통해 대량의 시료를 한 번에 분석할 수 있기 때문에 매우 획기적인 기술로 주목 받고 있다(Elshire et al., 2011). 이 후 보리와 밀, 콩 등을 이용한 GBS 분석이 빠른 속도로 확대되었으며(Poland et al., 2012; Sonah et al., 2013), 국내에서도 다양한 주요 작물을 대상으로 신품종 육성과 순계 선발, 유연 관계 분석 등의 유전체 연구에 이용되고 있다(Lee et al., 2016; Eun et al., 2016; Heo et al., 2017). 최근에는 포도에서도 전체 유전체 서열 분석과 GBS 기술을 이용한 SNP 대량 발굴을 통해 고밀도 유전자 지도를 작성하는 연구도 보고되었다(Hyma et al., 2015). 이와 같이 GBS 분석이 활발하게 이루어지고는 있지만, 염기서열 분석을 수행하기에 라이브러리의 크기가 다소 크거나 시료에 따라 증폭 양상이 특이하게 보이는 등의 매우 민감한 부분이 있어 효율적인 라이브러리를 제작하는데 큰 어려움을 겪고 있다.
본 논문은 과수에 대한 적용사례가 많지 않기 때문에 포도 작물을 이용하여 GBS분석 기술에 적용하고자 수행하였고, 염기서열 분석에 효율이 높은 GBS-라이브러리 제작 방법을 개선하였다. 이러한 결과는 포도뿐만 아니라 다른 과수작물 또는 다양한 수목에 대해서도 적용할 수 있을 것으로 기대한다.
Materials and Methods
공통 어댑터 및 96 바코드 어댑터 제작
본 연구에서 258점의 포도 유전자원을 한번에 빠르고 정확하게 분석하기 위한 일환으로 각 샘플을 구별하기 위해 96개의 서로 다른 바코드 어댑터 염기쌍을 Elshire et al. (2011)이 보고한 방법에 따라 제작하였다. 바코드 어댑터는 F-5’-ACACTCTTTCCCTACACGACGCTCTTCCGATCT xxxx-3’와 R-5’-CWGyyyyAGATCGGAAGAGC GTCGTGTAGGGAAAGAGTGT-3’두 가닥으로 구성하였다. 바코드 어댑터는 상보적인 33 염기와 서로 다른 4 - 7 염기(xxxx, yyyy)로 구성되어 있으며, CWG는 ApeKI 인식 자리로써 공통 어댑터-F와 상보적인 서열을 지니도록 제작하였다(Elshire et al., 2011). 공통 어댑터는 F-5’-CWGAGATCGGAAGAGCGGTTCAGCAGGAATGCCGAG-3’와 R-5’-CTCGGCATT CCTGCTGAACCGCTCTTCCGATCT-3’로 구성하였다. 두 가닥의 상보적인 서열로 구성된 어댑터는 gDNA에 ligation 시키기 전에 다음과 같이 annealing 과정을 수행한다. 1 × Elution buffer (EB, 10mM Tris - Cl, pH 8.0 - 8.5)를 이용하여 단일 가닥의 어댑터를 50 μM로 희석한 후, 각 어댑터 F와 L는 10 × Adapter buffer (AB, 500 mM NaCl, 100 mM Tris - Cl)를 넣어 10 μM로 만든다. PCR 기기를 사용하여 95℃에서 2분 동안 반응 후 25℃까지 0.1℃/s의 속도로 ramp down 시킨다. 그 다음에 25℃에서 30분 동안 반응하여 annealing 시키고 멸균수를 넣어 최종 2 μM의 농도로 희석하였다(Elshire et al., 2011).
효율적인 GBS 라이브러리 제작 방법
포도 유전자원의 고순도 gDNA 분리 및 제한 효소의 처리
국립원예특작과학원이 보유하고 있는 약 258점의 포도 유전자원을 제공받아 GBS 라이브러리 제작을 위한 재료로 사용하였다. 포도 유전자원으로부터 고순도의 genomic DNA를 분리하기 위해 258점의 포도 잎을 수집한 후 액화질소로 냉동시킨 다음 pestle을 이용하여 조직을 완전히 마쇄하였다. DNA의 순도를 높이기 위해 DNeasy Plant mini kit (Qiagen, USA)를 사용하여 매뉴얼에 따라 수행하였으며, RNase는 30분 동안 충분히 처리하여 RNA의 오염을 완전히 제거하였다.
이렇게 얻은 모든 DNA 샘플은 균일한 라이브러리 제작과 분포를 위해 SimplinanoTM spectrophotometer (GE Healthcare Life Sciences, UK)을 이용하여 샘플 당 100 ng으로 정량하였고, 정량화된 gDNA에 adapter를 붙이기 위해 제한효소(ApeKI, 3.6 U, NEB, England)를 넣어 총 20 μL에 맞춘 후 75℃에서 5시간동안 반응시켰다.
Adapter ligation, 각 샘플의 통합 및 정제
제한효소를 처리하여 절단된 DNA는 annealing시킨 바코드 어댑터와 공통 어댑터를 ligation하여 GBS 라이브러리를 증폭하기 위한 주형으로 하며, 절단된 DNA의 양 끝은 바코드 어댑터의 R - CWG (ApeKI 인식 자리)와 공통 어댑터의 F - CWG (ApeKI 인식 자리)가 각각 붙게 된다(Elshire et al., 2011). ApeKI 제한효소를 이용하여 절단된 DNA (20 μL)와 각각의 바코드 및 공통 어댑터(1 : 1)를 넣고, T4 ligase (Takara, Japan) 360U을 혼합하여 총 30 μL으로 조절한 후 22℃에서 2시간 동안 ligation 시켰다. 반응이 끝나면 65℃에서 30분 동안 처리하여 효소를 비활성화하였다. Ligation이 끝난 샘플은 각 5 μL씩 취하여 한 개의 e - tube에 혼합한 후, PCR 생성물의 순도를 높이기 위해 Qiaquick PCR purification kit (Qiagen, USA)를 이용하여 한 번 더 정제하였다.
고품질 라이브러리 제작
PCR 증폭에 사용된 프라이머는 F-5’-AATGATACGGCGACCACCGAGATCTACACTCTTTCCCTACACGAC GCTCTTCCGATCT-3’ (58 mer), R-5’-CAAGCAGAAGACGGCATACGAGATCGGTCTCGGCATTCCTGCTGA ACCGCTCTTCCGATCT-3’(61 mer)이다(Elshire et al., 2011). 프라이머 - F의 3’말단은 바코드 어댑터와 상보적으로 결합할 33개 염기서열을 포함하고 있고, 프라이머 - R의 3’말단은 공통 어댑터와 상보적으로 결합할 33개 염기서열을 포함하고 있다. PCR 증폭 반응을 위해 Accupower PCR premix kit (Bioneer, Korea)를 사용하였으며, 프라이머(10 pmol)는 프라이머-F/R 각각 1 μL, 증류수 43 μL를 넣어 최종 50 μL로 조절한 후 다음과 같은 조건으로 PCR을 수행하였다. 72℃ 5분; 98℃ 30초; [98℃ 30초; 65℃ 30초; 72℃ 20초] (16 cycles); 72℃ 5분의 조건으로 반응을 진행하였고, 완료된 라이브러리는 순도를 높이기 위해 Qiaquick PCR purification kit (Qiagen, USA)으로 한 번 더 정제하였다. 완료된 샘플은 겔 전기영동과 QC (quality control) 분석을 통해 최종적인 GBS 분석을 위한 적합 여부를 판단하고자 하였다.
염기서열 및 Genome-wide SNP 분석
염기서열 분석을 위해 Illumina Hiseq 2000을 이용하여 paired - end sequencing을 수행하였으며, 평균 101 bp의 reads가 생성되었다. SNP분석을 위해 바코드 시퀀스를 이용하여 demultiplexing을 수행한 후, Short read data의 sequence quality에 따른 trimming을 수행하였다(SolexaQA v.1.13). 분석된 전체 포도의 GBS 라이브러리의 공통 염기서열을 분석하기 위해 표준 유전체인 Vitis vinifera (Phytozome, 2017)의 염기서열을 이용하였으며, 258개의 포도 자원에서 분석한 염기서열과 표준 유전체의 염기서열을 이용하여 alignment (BWA)를 수행하였고 consensus sequence를 작성하였다(SAMtools v. 0.1.16). 분석 대상 간의 SNP 비교분석을 수행하기 위해 SEEDERS in-house script (SEEDERS Inc., Korea)를 이용하여 샘플 간 통합 SNP matrix를 작성하여 유의한 SNP후보를 선발하고(Kim et al., 2014), homo/heterogeneous type을 구분하여 대표 SNP를 선발하였다.
Results and Discussion
양질의GBS 라이브러리 제작
GBS 기술은 유전체 전체의 염기서열 분석 또는 염기서열 재분석(resequencing) 방식과 같은 NGS 기반 기술로서, 제한 효소와 바코드를 이용하여 대량의 시료를 한 번에 분석할 수 있으며, 다른 유전형 분석 기술과는 달리 저렴한 비용과 빠른 분석으로 높은 수준의 SNP 마커들을 탐색하는데 매우 용이하다. 또한, 이렇게 탐색된 SNP 마커들은 표준 유전체와 비교 후 염색체 상에 mapping 할 수 있다. 현재까지 보고된 많은 논문들은 채소, 식량 및 원예 작물에 집중되어 있으며, 과수나 수목에 대한 적용은 적은 편이다(Elshire et al., 2011; Poland et al., 2012; Hyma et al., 2015). 본 실험에서는 과수 작물 중 포도를 이용하여 GBS 라이브러리를 제작하고자 Elshire et al. (2011)에 의해 보고된 GBS 라이브러리 제작 방법을 이용하고자 하였으나, 이는 옥수수 또는 보리 유전자원을 통해 증명된 방법으로서, 포도에 적용시킨 결과 라이브러리의 품질이 균일하지 않았다. 따라서 본 연구에서는 기존에 보고된 GBS 라이브러리 제작 방법을 토대로 제한 효소의 사용량 및 처리 시간, 그리고 가장 중요한 PCR 증폭 조건 등을 개선하고 보완하였으며, 258점의 포도 유전자원을 이용하여 주요한 네 단계로 구분하여 GBS 분석을 위한 고품질의 라이브러리를 제작하였다.
첫째, 포도로부터 고품질의 gDNA를 분리하기 위해 기존의 방법을 이용할 수 있으나, 포도의 잎은 섬유질을 다량 함유하고 있기 때문에 고순도의 DNA를 얻기 어려운 경우가 종종 있으므로 column을 이용하여 분리하는 것을 권장한다. 또한, 포도 잎은 채집한 후에 즉시 DNA를 분리하는 것이 효율이 높았으며, 초저온 냉동고(-70℃)에 보관한 후(30 - 60일 이상)에 분리한 경우에는 효율이 다소 떨어지는 것을 확인하였다.
둘째, GBS 라이브러리를 제작하기 위해 분리한 DNA를 사용하여 제한 효소를 처리한다. 이 과정은 게놈 전체가 아닌 일부 염기서열만을 폭넓게 분석하기 위한 GBS 라이브러리 제작 과정 중 가장 중요한 단계라고 할 수 있다. 현재 Elshire et al. (2011)이 보고한 방법을 가장 이용하고 있는데, 포도의 경우 사용한 90% 이상의 포도 DNA가 이 방법과 같이 ApeKI을 2시간 동안 처리하였을 경우 10 - 20% 이상 잘리지 않은 DNA가 상당수 확인되었고, 섬유질을 많이 함유하고 있었던 포도 품종의 경우는 30%까지 제한 효소에 의해 잘리지 않음을 확인할 수 있었다(Fig. 1). 결과적으로 포도 시료를 이용한 제한 효소 처리는 기존에 알려진 시간 보다 최소 3시간 이상의 처리시간이 더 요구됨을 확인하였다.
셋째, 제한 효소의 충분한 처리가 이루어졌음에도 불구하고 각기 다른 96개의 바코드와 ligation시킨 샘플을 균일한 크기의 GBS 라이브러리로 증폭시키지 못한다면 좋은 결과를 얻을 수 없을 것이다. 높은 순도의 GBS 라이브러리는 어댑터 서열을 가지고 있는 DNA 단편(128 bp dimer 밴드)이 없고, 증폭된 DNA 단편이 170 - 350 bp일 경우에 GBS 분석이 매우 적합한 것으로 알려져 있다(Elshire et al., 2011). 또한, 현재 보편적으로 유전체 분석에 사용되는 플랫폼의 예로 Illumina사의 HiSeq2000은 100 - 400 bp 크기의 라이브러리 산물이 분석을 수행하기에 가장 적합하며, paired - end reads일 경우에는 가장 이상적인 크기를 250-500 bp로 권장하고 있다(Hamblin and Rabbi, 2014; Schroder et al., 2016). 이러한 보고는 기존의 방법을 사용했을 때 얻은 500 bp 이상 크기의 PCR 산물이 GBS 분석에 부적합하다는 것을 의미한다(Fig. 2A). 본 연구에서는 이를 극복하고 개선하기 위해 증폭된 DNA 단편의 길이가 200 - 400 bp 범위에서 주요 피크(main peak) 유지되도록 하기 위해 PCR을 이용한 증폭 단계 중 연장 단계(extension process, 72℃)의 시간을 기존의 방법(30초)보다 10초 줄인 20초로 단축시키고, 반복적인 cycle수를 최대 16회로 제한하였다. 그 결과 증폭된 DNA 단편의 크기가 500 bp 이하 수준으로 감소됨을 확인하였고, 증폭된 산물은 한 번 더 정제하여 최종적으로 남아있는 dimer를 완전히 제거함과 동시에 200 - 400 bp의 크기의 균일한 라이브러리 산물(DNA 단편)을 확보할 수 있었다(Fig. 2B).
Fig. 1.
Restriction enzyme digestion and multiplex enrichment PCR. (A) Restriction enzyme digestion. Modification of one enzyme (ApeKI) GBS protocol provides a uniform library for sequencing than the original protocol to grape. The 1, 2 lanes were treated for 2 hr by original protocol and the 3, 4 lanes were treated for 5 hr by improved method. (B) Multiplex enrichment PCR. The 1, 2 lanes were amplified using the original protocol (left). PCR products were purified (right). (C) The 3, 4 lanes were amplified using improved method (left), and purified (right). M: 1kb size marker.
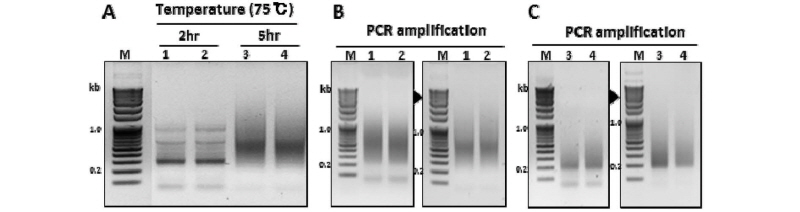
Fig. 2.
Evaluation of PCR fragment sizes in GBS library construction. Between 48 - 96 DNA samples were processed simultaneously. Appropriate primers on the ligated adapters are added and PCR is performed to increase the fragment pool. PCR products are cleaned up (A and C) and fragment sizes of the resulting library are checked on a DNA analyzer (Illumina Hiseq 2000, B and D). Improved libraries without adapter dimers are retained for DNA sequencing (C and D), but not original libraries (A and B). The frequency distribution of fragment sizes was evaluated to determine the proportion of fragments in the preferred size range (< 500 bp, blue arrows). The 1, 2 lanes were amplified using the original protocol (A). The 3, 4 lanes were amplified using improved method (C). M : 1kb size marker.
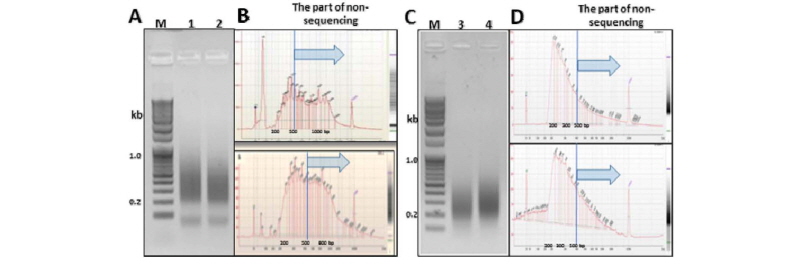
마지막으로, 위에서 언급한 최적화된 방법으로 제작된 최종 라이브러리 산물이 GBS 분석에 적합한지 여부를 확인하기 위해 QC (quality control) 분석을 수행하였다(Fig. 3). 기존 방법을 이용하여 제작한 라이브러리는 200 - 1,000 bp 범위로 매우 광범위한 크기로 분포하였고, 특히 400 - 600 bp 사이에 밀집되어 나타났다(Fig. 3A). 반면, 본 연구에서 제안한 방법을 이용하여 제작한 라이브러리 산물의 경우 200 - 500 bp 범위 내에 분포하였으며, 주로 200 - 400 bp에 밀집되어 나타남을 확인하였다(Fig. 3B). 이는 증폭된 DNA 단편이 170 - 350 bp일 경우에 GBS 분석이 매우 용이하다는 Elshire et al. (2011)의 연구 결과와 일치한다. 따라서 본 연구를 통해 제작한 라이브러리는 GBS 분석에 매우 적합한 것으로 판단되며, GBS를 통한 genome - wide SNP 분석과 다양한 유전체 분석에 매우 적절하게 활용될 것으로 기대된다(Oh and Jang, 2017).
Fig. 3.
The SNP distribution and density of mapped reads in 19 grape chromosomes. The distribution of total SNPs in 19 chromosomes of grape. Original method (A and B); (A) AAAACTT ‘B55 (GVIT0272), (B) TAGGAA ‘Battir’. Improved method (C and D), (C) GTCGATT ‘Cabernet Sauvignon’, (D) AGGC ‘Bellino’. The density was calculated as the average number of SNPs within a 500 kb region of each chromosome.

GBS 라이브러리를 이용한 SNP 추출 및 연관 분석
단일염기다형성(SNP)을 이용한 유전체 연구는 목표 형질을 조절하는 유전적인 요인을 결정하는데 유용하게 적용되고 있으며, 식량, 채소, 원예작물을 비롯하여 과수 작물인 포도에서도 다양한 SNP마커가 개발되고 있다(Adam-Blondon et al., 2004; Hyma et al., 2015; Hur et al., 2015). 본 연구에서는 위에서 언급한 두 가지 GBS 라이브러리 제작 방법을 비교하면서 SNP를 추출하기 위한 유전체 서열을 분석하였다. 이 때, 1차(66개 샘플)와 2차(48개 샘플)에 만든 라이브러리는 질(quality)이 매우 떨어져 염기서열을 두 번 반복하여 읽고 분석한 것이며, 이는 1차와 2차 시기에 사용한 GBS 라이브러리의 효율이 2배 이상 떨어진다는 것을 의미한다. 그럼에도 불구하고 단일 분석을 통해 얻은3차 라이브러리의 raw reads 수와 비슷한 결과를 나타냈고, 이는 최적화된 GBS 라이브러리를 만드는 것이 매우 중요하다는 것을 단적으로 보여준다. 또한, trimming과정을 통하여 얻은 1차와 2차 라이브러리 모두 95% 이상의 비율로 reads가 걸러져 3차와 4차 시기에 얻은 라이브러리에 비해 상대적으로 높았으나, 최종적으로 게놈 내에 mapping되어 분석에 사용되는 reads 수의 비율은 1차와 2차의 평균은 71.7%이고, 3차와 4차는 평균 81.87%를 나타냈다. 따라서 본 연구에서 개선된 방법을 통해 제작한 GBS 라이브러리가 mapping 효율을 10.17% 더 증가시켰다는 것을 알 수 있었다(Table 1).
|
Table 1. Summary of sequencing data and reads obtained from SNPs of GBS library against grape reference genome. 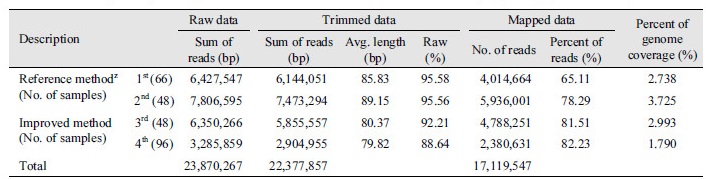
|
|
|
zElshire et al., 2011. |
|
Mapping 효율과 관련하여 각 염색체 내 SNP의 분포도를 조사하였고, GBS 라이브러리를 제작한 시기별로 각각 하나의 샘플을 무작위로 선발하였다(Fig. 3). 그 결과, 상대적으로 1차와 2차 라이브러리의 depth가 높을 뿐 모든 라이브러리 샘플에서 포도 19개의 염색체 내에 mapped reads가 균일하게 분포되어 있는 것을 확인하였다. 더불어 4차 라이브러리는 96개 시료를 한 번에 통합하여 정제했음에도 불구하고 염기서열의 질이 좋아 한 번에 분석 가능하였고, 3차(48개 샘플) 라이브러리를 분석한 것보다 reads 수는 적으나 mapping율은 유사하게 나타난 것으로 보아 본 연구에서 개선시킨 GBS 라이브러리 구축 방법을 이용하면 분석하고자 하는 샘플의 수가 많은 경우에도 효율이 유지되거나 증가함을 확인할 수 있었다.
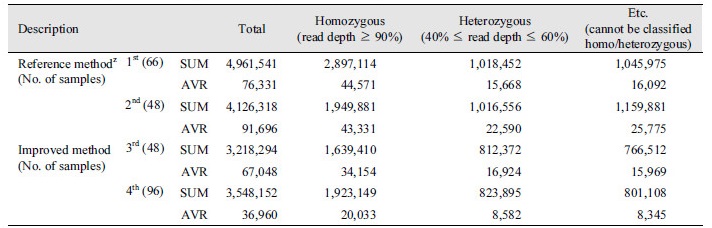
이를 바탕으로 mapped reads간의 SNP 비교분석을 수행하기 위해 SEEDERS in-house script를 이용하여 샘플 간 통합 SNP matrix를 작성하였다(Kim et al., 2014). SNP는 read depth를 기준으로 ≥ 90%일 경우 homo type, 40% ≤ 이면서 ≤ 60%일 경우 heterogeneous type으로 구분하여 대표 SNP를 선발하였고, 그 외 범주에 해당되는 SNP는 homo/heterogeneous type으로 구분할 수 없는 경우로 구분하였다(Table 2). 앞서 설명한 것과 같이 두 번을 반복하여 읽은 영향으로 1, 2차의 전체 SNP의 수가 높은 것으로 나타났으나 모든 실험군에서 전체 SNP수에 비례하여 약 50% 이상 homo, 약25%는 hetero 그리고 나머지는 기타 범주로 구분되었다. 따라서 본 방법 역시 과수작물인 96개의 포도 샘플에 대해서도 단 한번의 분석으로도 두 배에 가까운 높은 효율을 얻을 수 있었다.
Conclusion
GBS 기술은 유전체 전체의 염기서열 분석 또는 염기서열 재분석방법과 같은 NGS 기반 기술로서 적은 비용으로 대량의 샘플을 분석할 수 있는 장점을 가지고 있다. 이를 위해GBS 라이브러리를 잘 구축하는 것이 매우 중요하며, 이미 옥수수와 벼, 보리, 콩 등의 작물에서 활용되고 있다. 또한, 포도와 같은 과수작물에서도 활용되고 있으나 기존의 방법으로는 다소 효율이 떨어지는 단점이 있기 때문에 본 연구에서 효율을 개선시키고자 하였다. 결과적으로 본 실험에서 기존에 사용하고 있던 제한 효소의 사용량(ApeKI, 3.6 U, NEB, England) 및 처리 시간(75℃ 5시간 이상), PCR 증폭 조건(Extension 시간; 20초 단축, 증폭cycle; 16회이내로 단축)등을 개선하여 라이브러리의 크기를 200 - 400 bp로 균일하게 제작할 수 있도록 방법을 확립하였다. 이는 기존의 방법을 사용했을 때보다 약 10.2%의 mapping 효율을 증가시켰을 뿐만 아니라, 96개 샘플을 통합한 경우에도 mapped reads가 포도의 19개 염색체 내에 고르게 분포함을 확인하였다. 즉, 개선된 GBS 라이브러리 제작을 이용하여 과수 작물 및 수목에도 균일한 GBS 분석용 라이브러리 제작 및 유전체 분석의 효율을 높일 수 있을 것으로 기대된다.

